A couple of structures
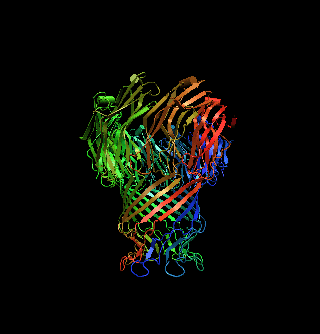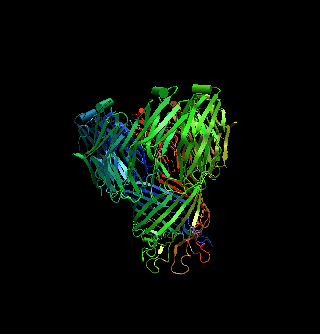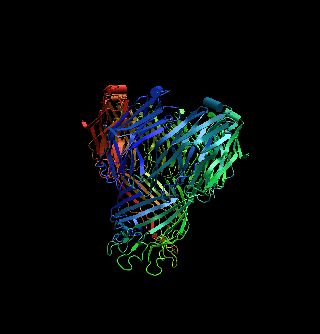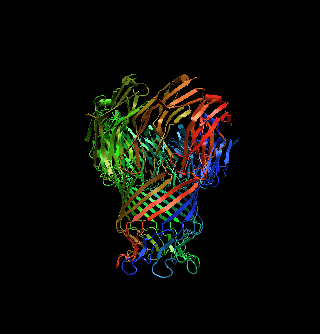 |
| The mycobacterial porin |
|
|
Movie of the structural changes during a catalytic cycle of nucleoside monophosphate kinasesClemens Vonrhein, G. J. Schlauderer and Georg E. Schulz (1995). Structure 3, 483-490 There are 17 crystal structures of nucleoside monophosphate kinases known. As expected for kinases, they show large conformational changes upon binding of substrates. These are concentrated in two chain segments, or domains, of 30 and 38 residues that are involved in binding of the substrates N1TP and N2MP (nucleoside tri- and monophosphates with bases N1 and N2), respectively. After aligning the 17 structures on the main parts of their polypeptide chains, two domains in various conformational states were revealed. These states were caused by bound substrate (or analogues) and by crystal-packing forces, and ranged between a 'closed' conformation and a less well defined 'open' conformation. The structures were visually sorted yielding an approximately evenly spaced series of domain states that outlines the closing motions when the substrates bind. The packing forces in the crystals are weak, leaving the natural domain trajectories essentially intact. Packing is necessary, however, to produce stable intermediates. The ordered experimental structures were then recorded as still pictures of a movie and animated to represent the motions of the molecule during a catalytic cycle. The motions were smoothed out by adding interpolated structures to the observed ones. Given the proliferating number of homologous proteins known to exist in different conformational states, it is becoming possible to outline the motions of chain segments and combine them into a movie, which can then represent protein action much more effectively than static pictures alone are able to do. For further information and movie downloads Enter Here |
|
Crystal structure of 6-hydroxy-D-nicotine oxidase from Arthrobacter nicotinovoransJochen, W. A. Koetter and Georg E. Schulz (2005). J. Mol. Biol. 352, 418-428 The crystal structure of 6-hydroxy-D-nicotine oxidase was solved by X-ray diffraction analysis in three crystal forms at resolutions up to 1.9 A. The enzyme is monomeric in solution and also in the mother liquor but formed disulfide-dimers in all crystals. It belongs to the p-cresol methylhydroxylase - vanillyl-alcohol oxidase family and contains an FAD covalently bound to the polypeptide. The covalent bond of this enzyme was the first for which a purely autocatalytic formation had been shown. In contrast to previous reports, the bond does not involve N(epsilon2) (N3) of His72 but the N(delta1) (N1) atom. The geometry of this reaction is proposed and the autoflavinylation is discussed in the light of homologous structures. The enzyme is specific for 6-hydroxy-d-nicotine and is inhibited by the l-enantiomer. This observation was verified by modeling enzyme-substrate and enzyme-inhibitor complexes, which also showed the geometry of the catalyzed reaction. The binding models indicated that the deprotonation of the substrate rather than the hydride transfer is the specificity-determining step. The functionally closely related 6-hydroxy-l-nicotine oxidase processing the l-enantiomer is sequence-related to the greater glutathione reductase family with quite a different chainfold. A model of this "sister enzyme" derived from known homologous structures suggests that the reported l-substrate specificity and d-enantiomer inhibition are also determined by the location of the deprotonating base. PDB entry: 2BVF. |
Click to enlarge
|
|
Click to enlarge
|
The structure of a retinal-forming carotenoid oxygenaseKloer, D. P., Ruch, S., Al-Babili, S., Beyer, P. and Schulz, G. E. (2005). Science 308, 267-269 Enzymes that produce retinal and related apocarotenoids constitute a sequence- and thus structure-related family, a member of which was analyzed by x-ray diffraction. This member is an oxygenase and contains an Fe2+-4-His arrangement at the axis of a seven-bladed ß-propeller chain fold covered by a dome formed by six large loops. The Fe2+ is accessible through a long nonpolar tunnel that holds a carotenoid derivative in one of the crystals. On binding, three consecutive double bonds of this carotenoid changed from a straight all-trans to a cranked cis-trans-cis conformation. The remaining trans bond is located at the dioxygen-ligated Fe2+ and cleaved by oxygen. PDB entry: 2BIW. |
The structure of a mycobacterial outer-membrane channelMichael Faller, Michael Niederweis and Georg E. Schulz (2004). Science 303, 1189-1192 Mycobacteria have low-permeability outer membranes that render them resistant to most antibiotics. Hydrophilic nutrients can enter by way of transmembrane-channel proteins called porins. The structure of the main porin from Mycobacterium smegmatis, MspA, revealed a homooctameric goblet-like conformation with a single central channel. This is the first structure of a mycobacterial outer-membrane protein. No structure-related protein was found in the Protein Data Bank. MspA contains two consecutive ß barrels with nonpolar outer surfaces that form a ribbon around the porin, which is too narrow to fit the thickness of the mycobacterial outer membrane in contemporary models. PDB entry: 1UUN. |
Click to enlarge
|
|
Click to enlarge
|
Structure and action of urocanaseDirk Kessler, J. Retey and G. E. Schulz (2004). J. Mol. Biol. 342, 183-194 Urocanase is a symmetric homodimer of 2 x 557 amino acid residues with tightly bound NAD+ cofactors. Each subunit consists of a typical NAD-binding domain inserted into a larger core domain that forms the dimer interface. The core domain has a novel chain fold and accommodates the substrate urocanate in a surface depression. The NAD domain sits like a lid on the core domain depression and points with the nicotinamide group to the substrate. Substrate, nicotinamide and five water molecules are completely sequestered in a cavity. Most likely, one of these water molecules hydrates the substrate during catalysis. This cavity has to open for substrate passage, which probably means lifting the NAD domain. The observed atomic arrangement at the active center gives rise to a detailed proposal for the catalytic mechanism that is consistent with published chemical data. As expected, the variability of the residues involved is low, as derived from a family of 58 proteins annotated as urocanases in the data banks. However, one well-embedded member of this family showed a significant deviation at the active center indicating an incorrect annotation. PDB entry: 1UKW. |
Conversion of squalene to the pentacarbocyclic hopeneDirk J. Reinert, Gianni Balliano and Georg E. Schulz (2004). Chem. Biol. 11, 121-126. The membrane protein squalene-hopene cyclase was cocrystallized with 2-azasqualene and analyzed by X-ray diffraction to 2.13 Å resolution. The conformation of this close analog was clearly established, and it agreed with the common textbook presentation. The bound squalene undergoes only small conformational changes during the formation of rings A through D, thus requiring no intermediate. However, ring E formation is hindered by an entropic barrier, which may explain its absence in the steroids. The structure analysis revealed a mobile region between the active center cavity and the membrane, which may melt, opening a passage for squalene and hopene. PDB entry: 1UMP. |
Click to enlarge
|